
The Lowen Lab uses molecular tools, classical virology, and both human and animal models of infection to study the biology of RNA viruses, with a focus on influenza A virus. We are particularly interested in features of viral biology that shape evolutionary dynamics and the mechanisms governing transmission between individuals.
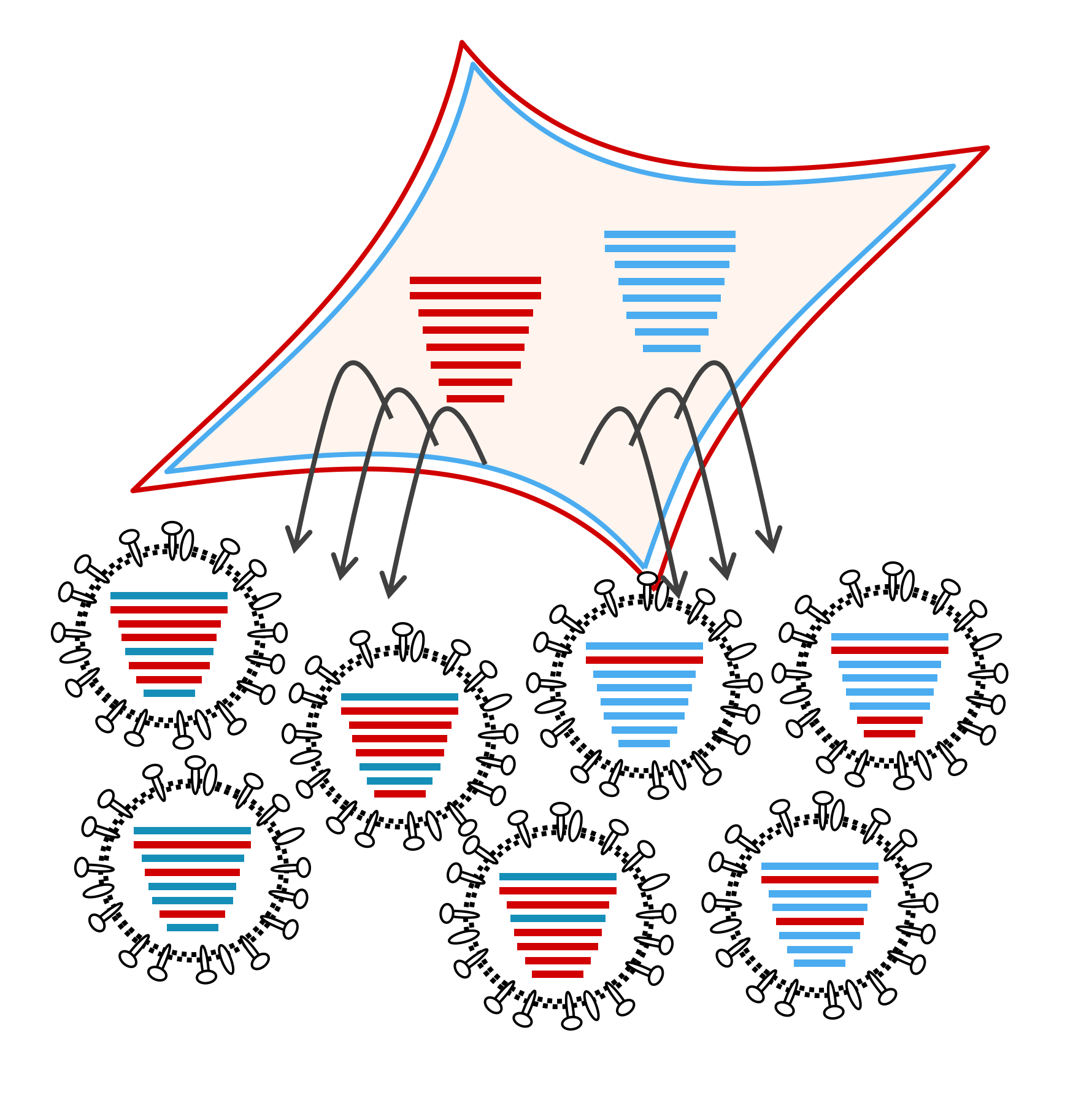
Much of our prior and current work focuses on reassortment – a mechanism by which co-infecting viruses exchange genetic information. In broader terms, we are interested in interactions among co-infecting viruses and the implications of these interactions for viral propagation and evolution.
A major current focus seeks to understand the features of influenza A virus biology that support and constrain adaptation to new environments, including new immune pressures or new host species. This work involves evaluation of viral population dynamics within infected hosts and between hosts during transmission and employs both experimental systems and samples taken from naturally infected individuals.
We are also actively working to understand the fundamental question of how influenza A viruses transmit and how modes of transmission vary with host, viral and environmental features. This work is geared to enable the evidence-based design of interventions that can limit transmission.